


En los albores de la ciencia
El médico y paleontólogo norteamericano Marvin J. Allison (1921 – 2015), famoso por sus estudios en Chile y descubridor de la existencia de la tuberculosis en la América precolombina, escribió en 1979: «El conocimiento de la historia de una enfermedad indica el camino para reducirla o erradicarla».
La historia de la fibrosis quística (FQ) es tan vieja como la del hombre. Hasta su reconocimiento como entidad propia por la comunidad médica, los fallecimientos por FQ fueron atribuidos a otras causas, y todavía lo pueden ser en países no desarrollados, por lo que conocer el número de fibróticos quísticos en el mundo sigue siendo, a día de hoy, materia de especulación; por otro lado, la ausencia de diagnóstico y tratamiento adecuado sin duda hace difícil su supervivencia y el número de portadores puede continuar en crecimiento.
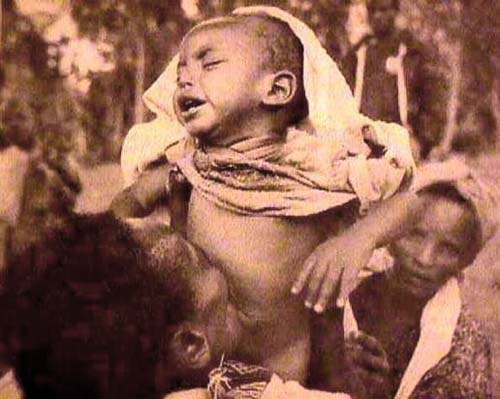
Figura 1. Brujo lamiendo a un niño
Es difícil establecer el punto inicial de la FQ, ya que al tratarse de una enfermedad genética es posible que haya estado siempre presente desde que el hombre es. X. Estivill estima que pudo aparecer en Europa hace unos 52.000 años. Mucho antes de que se reconociese como entidad patológica, existían observaciones recogidas en el antiguo folclore popular del norte de Europa en las que se decía que los niños que al besarlos tenían sabor salado, estaban embrujados y morirían precozmente (Figura 1). En un manuscrito alemán del siglo XV (Codex Latinus Monacensis 849), -realmente un grimorio o manual de nigromancia y artes adivinatorias escrito en latín- se recoge la bendición Wider elbe contra las enfermedades de los niños encantados; dicho códice recomienda lamer la nariz del niño, supuestamente encantado, para averiguar si tiene o no sabor salado –so sint es dy elbe-. Es la primera documentación escrita que relaciona el sabor salado con una posible enfermedad, relación que hoy es ampliamente conocida con la FQ.
En el año 1606 aparece en España una descripción relacionada con este tema; en la literatura médica de nuestro Siglo de Oro, Juan Alonso y de los Ruices de Fontecha, más conocido como Juan Alonso de Fontecha (Daimiel, Ciudad Real, 1560 – Alcalá de Henares,1620) médico, obstetra, farmacólogo, escritor español y profesor de medicina de la Universidad de Alcalá, en su libro “Diez privilegios para mugeres prenadas” dice: “… que se conoce a la gente embrujada, si al rascarles en la frente, uno después nota un sabor salado en los dedos” (Figura 2). Otras referencias al sabor a sal y su relación con el encantamiento de los niños se dan en la literatura europea del siglo XVII.

Figura 2. Portada de ‘Diez privilegios para mugeres prenadas’
Probablemente la primera descripción patológica macroscópica del proceso se debe al anatomista y botánico holandés Peter Paaw (1564 – 1617), quién en 1595 en Leiden, universidad de la que fue rector, realizó la autopsia de una niña de once años y escribe: “… el páncreas estaba abultado, cirroso y de color blanco brillante después de cortarlo y abrirlo…”; “… La causa de muerte fue el páncreas.”, concluye Paaw. Vemos aquí la relación entre la superstición o hechizo y la causa orgánica de la enfermedad que nos ocupa. También de Holanda es la anotación que Gerardus Leonardus Blasius (1627 – 1682), anatomista, escribe en su libro de observaciones raras de 1677 sobre el páncreas cirroso de un niño de nueve años a quien estudió.
Una de las primeras “Historias clínicas” fue la realizada por Georg Seger (Nuremberg ,1629 – 1678) en Thorun, Polonia, en 1673; refiriéndose a una niña de tres años de edad, recoge la presencia de fiebre, vómitos, diarrea, dificultades para ganar peso e inanición prolongada. La autopsia de la pequeña paciente -practicada en pleno auge de las conocidas como Gimnasium Anatomicum– mostró un páncreas endurecido y cirroso.
Nils Rosén von Rosenstein (1706, Sexdrega – 1773, Uppsala), eminente pediatra sueco y profesor en Uppsala desde 1740, coincidiendo allí con C. Linnaeus, en su libro sobre enfermedades infantiles de 1764 describe un cuadro, dentro del apartado genérico de los procesos diarreicos, al que denomina “fluxus coeliasus”, que se caracterizaba por la presencia de diarrea, distrofia, falta de medro y debilidad; pese a ello los enfermos tenían un apetito voraz. Describe además dilataciones en manos y pies y un vientre distendido con páncreas endurecido. Probablemente eran en su mayoría enfermos con FQ. También, entre otras novedades, introdujo en Suecia el uso de la quinina y la variolización contra la viruela. Los trabajos de N. Rosén fueron ampliamente traducidos y su obra traducida al inglés –The Diseases of Children and their Remedies (1776)– se puede considerar como el primer tratado de pediatría de amplia difusión.
Carl Von Rokitansky (1804, Hradec Králové, Bohemia – 1878, Viena) describe -en la pujante Viena de 1838- los resultados de una de las miles de autopsias por él practicadas: se trataba de un feto de siete meses de gestación, sin signos de vida postnatal, en el que se detectó una perforación de intestino delgado y flujo de meconio libre en la cavidad peritoneal con reacción inflamatoria. Presumiblemente se trataba de lo que hoy conocemos como íleo meconial. Rokitansky, fundador y promotor de la conocida como Segunda Escuela de Viena, enfocada bajo la óptica del método científico, desarrolló un método de autopsia conocida con el epónimo ‘Técnica de Rokitansky’, que es todavía uno de los métodos estándar empleados hoy en día, basada en el examen in situ de las vísceras.
En 1850, Alois Bednar (1816, Potterstein, Bohemia – 1888, Viena) pediatra austriaco, jefe médico del Hospital de Niños Expósitos de Viena y profesor asociado de su universidad, aplicando las técnicas de Rokitansky a la patología infantil describe un caso similar en un recién nacido vivo que murió al sexto día de vida. Más o menos coincidentes en el tiempo se describen y publican cuadros similares en Inglaterra.
Siglo XX y …
Existe controversia sobre a quién atribuir la primera descripción de la fibrosis quística en los tiempos modernos. Siguiendo a D.A. Christie y E.M. Tansey la que podríamos considerar como la primera descripción científica de la fibrosis quística en el siglo XX podría ser atribuida a Blackfan y Wolbach (1933), si bien la existencia de alteraciones pancreáticas y bronquiectasias en niños autopsiados no fue relacionada con otra cosa que no fuese un déficit de vitamina A. Sí lo hizo el Prof. Guido Fanconi quien, en 1936, publica un artículo titulado Das Coeliacsyndrom bei Angeborener zysticher pankreas fibromatose und bronchiektasen en el que establecía la relación entre enfermedad celiaca, fibrosis quística del páncreas y bronquiectasias; y que el denominaba “fibromatosis quística pancreática familiar con bronquiectasias”. G. Fanconi (1892, Poschiavo, Suiza – 1979, Zurich) desarrolló toda su carrera como pediatra hospitalario en el Kinderhospital de Zurich; en 1934 el reconocido, al menos retrospectivamente, como primer caso de fibrosis quística del páncreas fue descrito en una tesis bajo su dirección y, casi una década antes –1928– publicó observaciones concernientes al denominado por él “síndrome celiaco” en un grupo de niños con síntomas digestivos desde la lactancia y patología respiratoria asociada.

Figura 3. Dorothy Hansine Andersen (a)
La primera correlación descriptiva entre la clínica e histopatología de la fibrosis quística como entidad propia se debe a Dorothy Andersen, (Dorothy Hansine Andersen,1901, Asheville, North Caroline – 1963, New York) (Figura 3) patóloga del Columbia Presbyterian Medical Center de Nueva York y profesora de su prestigiosa universidad homónima, quien comunicó en mayo (American Pediatric Society) y publicó en agosto de 1938 una detallada revisión de los signos de esta enfermedad, incluyendo la asociación con el íleo meconial y denominándola “cystic fibrosis of the pancreas” : “ … The diagnosis of cystic fibrosis of the pancreas can be made with certainty only by examining the duodenal contents for pancreatic enzymes or by microscopic examination of the pancreas”; si bien lo relacionaba con deficiencias de vitamina A; teoría que sostuvo durante muchos años sin la evidencia científica suficiente. Su trabajo se realizó sobre los pacientes fallecidos en el Babies’ Hospital de dicho centro. Al año siguiente se hizo el primer diagnóstico in vivo de un paciente con fibrosis quística, diagnóstico complejo para paciente y médicos ya que se basó en la cuantificación de enzimas pancreáticos en la secreción duodenal por colocación de sondaje duodenal. En aquellos años, el diagnóstico se realizaba por la existencia de antecedentes familiares, insuficiencia pancreática y afectación pulmonar crónica.
Para 1944 ya era claro para unos pocos médicos interesados que la fibrosis quística era una enfermedad no confinada al páncreas, sino que afectaba a otros órganos con secreción exocrina como los pulmones. Por tanto, el nombre no parecía totalmente apropiado para este cuadro y Sydney Farber (1903, Búfalo – 1973, Boston), profesor de patología en la Harvard Medical School de Boston la denominó “mucoviscidosis”, en base a los hallazgos observados en las autopsias de pacientes fallecidos por lo que se empezaba a denominar fibrosis quística y que el definió como una enfermedad sistémica y no de un órgano concreto.
A partir de aquí parecía evidente que la mucoviscidosis bronquial tenía mucho que ver en la patogenia de la FQ. Una afortunada coincidencia médica fue el desarrollo a finales de la década de los cuarenta de nuevos antibióticos a partir del descubrimiento de la penicilina por Sir Alexander Fleming en 1929, trabajando en el St Mary’s Hospital de Londres. Fleming descubrió la penicilina, pero esta no estuvo disponible para su uso clínico, y con restricciones por las dificultades de producirla masivamente, hasta 1941.
Andersen y Hodges (1946) del estudio de familias con pacientes que padecían esta enfermedad, concluyeron que la incidencia familiar observada era concordante con una herencia autosómica recesiva. Si bien Andersen continuaba creyendo que “the pulmonary infection is the result of the nutritional deficiency”; esta era la primera vez que se establecía de una manera clara, basada en evidencias clínicas, que la FQ era una enfermedad recesiva según patrones mendelianos.
Bodian, en 1952, elaboró la teoría patogénica de que las lesiones que observaban en el páncreas, pulmón, hígado y conductos deferentes se debían a secreciones anormalmente espesas que taponaban los conductos excretores de las glándulas exocrinas, produciéndose secundariamente la dilatación quística, fibrosis y destrucción de la glándula; este autor describió por primera vez la cirrosis biliar focal, lesión patognomónica de la FQ en el hígado.
1953 marcó un hito en el conocimiento de la FQ con la detección del defecto en las glándulas sudoríparas por Paul di Sant´Agnese. Di Sant’Agnese (1914 – 2005) – junto a Harry Shwachman y Dorothy Andersen – fue fundador de la Cystic Fibrosis Foundation and of Cystic Fibrosis Care en los Estados Unidos, desde el Columbia University Medical Center de Nueva York. En 1946 había sido también el primero en usar la penicilina inhalada para el tratamiento de la FQ. En el año 1949 se produjo una ola de calor que originó que muchos pacientes con FQ, especialmente lactantes, sufrieran deshidrataciones con alcalosis hipoclorémica y postración debido a la pérdida de sales; Di Sant’Agnese –patólogo en el equipo de Andersen- investigando la causa de estas pérdidas excesivas, llegó a la conclusión de que se debían a la eliminación anormal de cloro por el sudor. Esto le llevó a desarrollar –1952- el que sigue siendo test diagnostico prínceps para la FQ, el test que cuantifica electrolitos en sudor –sweat test –y que abrió la puerta a las investigaciones para identificar el defecto básico de la enfermedad. Antes de descubrir este test, la FQ se diagnosticaba por medio de la intubación duodenal para demostrar la insuficiencia pancreática que acompañaba al cuadro. La determinación de sodio en sudor se convirtió en el mejor método diagnóstico de la FQ. Inicialmente se sometía a los pacientes a altas temperaturas para inducir la sudoración, lo cual no estaba exento de riesgos, pero en 1959, mediante el test de iontoforesis con pilocarpina diseñado por Gibson y Cook, se pudo realizar de forma segura, siendo hasta la fecha una prueba diagnóstica no superada. Lewis E. Gibson y Robert E. Cooke del Department of Pediatrics, Johns Hopkins Medical School, Johns Hopkins University publicaron sus conclusiones en 1959. Gibson (1927 – 2008) se incorporó al equipo de R. Cooke en la Johns Hopkins y ulteriormente fue director de diversos grupos de investigación en FQ hasta su retiro como profesor de pediatría en la Loyola University de Chicago y director de su centro de FQ en 1996. Robert Edmond Cooke (1920-2014) fue un pediatra profundamente implicado en la reforma de la sanidad americana durante las administraciones de John F. Kennedy –de quien fue estrecho colaborador- y Lyndon B. Johnson.
Durante la década de los años 50-60, aunque la causa fundamental de lesión se desconocía, se fueron perfilando las diferentes formas clínicas. Shwachman (Boston, 1910 – 1986) fue el primero en publicar que el 15% de los pacientes no tenían afectación pancreática; y estableció un sistema de puntuación clínico de la gravedad que todavía hoy se sigue utilizando.

Figura 4. Paul M. Quinton (b)
Los recién nacidos con íleo meconial tenían un pronóstico sombrío: cerca del 50% fallecían. La realización y publicación -1957, The Children’s Hospital of Philadelphia – de una técnica denominada ileostomía por Bishop y Koop, permitió salvar muchas vidas. Una década más tarde, 1969, Helen Noblett (Royal Children’s Hospital, Melbourne) utilizó enemas de amidotrizoato meglumina en el íleo meconial no complicado, lo que consiguió la curación de los niños sin realizar procedimientos quirúrgicos.
Otros avances en el campo de la farmacología, con la aparición de penicilinas resistentes a betalactamasas y la introducción de fermentos pancreáticos con cubierta entérica, contribuyeron de manera decisiva a la supervivencia de estos enfermos.
El patrón de herencia autosómico recesivo de la FQ fue claramente indicado por el grupo de Harvard en 1956 (Steinberg, Allen, Shwachman and Dooley), siendo Fred Allen su genetista y confirmado por P.M. Conneally, ya en 1973.
En la década de los 80 los científicos descubrieron que el mal funcionamiento de los tejidos epiteliales era un defecto común a todos los órganos afectados por la FQ. En particular se reveló que los epitelios de los pacientes eran impermeables al ión cloruro (M. Knowles,1981), (P.M. Quinton, 1983) (Figura 4). Quinton (Universidad de California) explicó el porqué del sudor salado en los pacientes con FQ. La incapacidad del tejido epitelial de absorber cloruro y la consiguiente imposibilidad de absorción del sodio causa la excesiva retención de estos iones en el sudor y lo convierte en anormalmente salado; el descubrimiento de que el defecto específico de la FQ era una reabsorción defectuosa del cloro a nivel de las células epiteliales del epitelio glandular marcó un cambio en la investigación de esta enfermedad.

Figura 5. Francis Collins (c)
En 1985 se localizó el gen en el cromosoma 7 (L. C. Tsui, 1985) y a finales de esta misma década quedó patente que la FQ se debía al mal funcionamiento de un canal de cloruro, hecho que se confirmó con la identificación del gen y su producto proteico (CFTR o Cystic Fibrosis Transmembrane Regulator) en 1989 por parte de Lap-Chee Tsui y John Riordan (Toronto) junto con Francis S. Collins (Michigan) (Figura 5). En 1991, se demostró que la proteína CFTR forma un canal de cloruro.
Desde la identificación del gen se han abierto nuevas vías de investigación que incluyen la obtención de ratones transgénicos en 1992, estudios mutacionales y análisis funcionales de la proteína CFTR en células epiteliales. Cabe destacar que los distintos modelos de FQ murinos tienen patología intestinal pero no desarrollan alteraciones respiratorias. Sin embargo, desde 1997, existen dos cepas de ratones que sí presentan patología respiratoria. En 1997, G. Kent y colaboradores describieron el fenotipo de una cepa de ratones que desarrollan patología pulmonar espontánea y progresiva, de aparición temprana, con características de fibrosis e inflamación y problemas en el transporte mucociliar. Más recientemente, se ha generado un ratón con patología pulmonar muy semejante a la fibrosis quística (M. Mall, 2004). El transporte defectuoso de moco causó una patología pulmonar severa y espontánea comparable a la FQ, con inflamación, obstrucción por moco y poca eliminación bacteriana por lo que estos ratones constituyen un mejor modelo para el estudio de la afectación pulmonar en la FQ.
Hoy la ciencia marca ya el camino que la hechicería inició, pero si lo pensamos bien, el fabricar ratones que padezcan enfermedades humanas podía habernos conducido a la hoguera en el siglo XVI … y besar la frente de un recién nacido y deducir qué hay tras el sabor salado que notamos en los labios puede ser un empirismo científico que nos viene de aquellos tiempos. Hechiceros y médicos suelen ir de la mano. Y más si son pediatras. ¿O no?
Javier Pérez-Frías (1,2,3), María del Carmen López Castillo (3), Raquel Yahyaoui (2,3)
1.Universidad de Málaga; 2.IBIMA; 3.Hospital Regional Universitario de Málaga
Referencias de las imágenes
a. Dorothy H. Andersen (1901-1963; Carolina del Norte, EE.UU). National Library of Medicine. Wikimedia Commons, el repositorio multimedia libre. [Public domain]. https://www.nlm.nih.gov/changingthefaceofmedicine/physicians/biography_8.html
b. Paul M. Quinton. From www.biomed.ucr.edu website.
c. Francis Collins. Official portrait of NIH [Public domain]. Attribution: Bill Branson. https://commons.wikimedia.org/wiki/File:Francis_Collins_official_portrait.jpg